
New FDA QMSR Through The Lens Of Risk Management: Requirements And Analysis
By Edwin Bills, Christie Johnson, and Jayet Moon

On Feb. 2, 2024, the FDA set a new cornerstone in the medical device regulatory landscape by releasing the new Quality Management System Regulation (QMSR), a forward step that is scheduled to be fully implemented by Feb. 2, 2026. This regulation marks a pivotal transition from the guidelines previously stated in 21 CFR 820.5, forging a pathway toward an integrated approach that aligns with global standards. Now, under QMSR, Clause 4.1.1 of ISO 13485 requires organizations to document a QMS and maintain its effectiveness in accordance with the requirements of the standard and international regulations.
This new regulation is not simply procedural; it is a foundational requirement since, ultimately, we are here to provide a system to help patients that is safe and effective.
This article follows up on our May 24, 2023, Med Device Online article, The Intersection of ISO 13485 and ISO 14971 Under the Proposed FDA QMSR.
Evolution Of The Regulation
The following timeline traces the evolution of quality management systems through history:
- 1978: Introduction of Good Manufacturing Practices (GMP) — The FDA established GMP regulations for medical devices, aiming to ensure their quality and safety.
- 1994: Draft Proposal and Public Comments — A draft of the QSR was proposed in 1994, which received significant attention from the public and stakeholders. Approximately 10,000 public comments were submitted, reflecting widespread interest and engagement in the regulatory process.
- 1996: Introduction of QSR 21 CFR 820 — Building upon the foundation laid by the GMP regulations, the FDA introduced the Quality System Regulation (QSR) outlined in 21 CFR 820. This update modernized and expanded upon the regulatory framework, aligning it more closely with international standards (ISO 9001) and best practices. The QSR incorporated principles and requirements from ISO 9001:1994 and the Committee Draft (CD) of ISO 13485, aligning the FDA's regulations more closely with international quality management standards.
- Stability Since 1996 — Since its introduction in 1996, the QSR has remained relatively stable, with no significant revisions made to its core principles and requirements. However, there have been updates and additions to address emerging issues such as unique device identification (UDI) and combination products.
- Evolution of ISO 13485 — While the QSR has remained largely unchanged, ISO 13485, the international standard for medical device quality management systems, has undergone two revisions since 1996. These revisions have aimed to enhance the effectiveness and relevance of the standard, including a shift toward a process-oriented model.
- Divergence from ISO 9001:2015 — Despite the evolution of ISO standards, particularly ISO 9001 transitioning to a high-level structure in 2015, ISO 13485 has not adopted this model. The focus remains on maintaining a process-based approach tailored specifically to the medical device industry.
- QMSR — The proposed QMSR received less attention compared to the 1994 draft of the QSR, with fewer than 100 public comments submitted. This suggests a shift in stakeholder engagement or perhaps greater satisfaction with the existing regulatory framework. This regulation adopts ISO 13485:2016 in large part by reference and greatly simplifies compliance.
What Does QMSR Do To The Existing QSR?
In the updated regulatory landscape, the new QMSR withdraws most of the requirements in the previous Part 820 but preserves the scope and certain definitions integral to the Quality System Regulation.
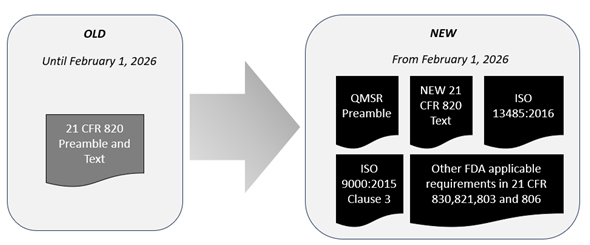
The QMSR incorporates by reference ISO 13485:2016 with selectively adopted provisions to ensure consistency with other applicable FDA requirements. Moreover, it adopts Clause 3 of ISO 9000:2015, which defines the essential terminology for applying ISO 13485 standards effectively.
This revision extends to include edits to Part 4 (cGMPs) for combination products while maintaining the cGMP requirements.
To fully understand the QMSR’s nuances and underlying intentions, a thorough examination of the preamble and responses to comments is crucial. The figure above shows that manufacturers must now reference multiple documents for a comprehensive gap analysis rather than just one consolidated text. In this vein, we will analyze the important comments in the preamble associated with risk management in this article.
Structure Of New Part 820
FDA is incorporating by reference the current 2016 version of ISO 13485 and the current 2015 version of Clause 3 of ISO 9000. This incorporation has led to the FDA reserving many sections of the old Part 820. Readers should pay special attention to sections that have not been reserved to check for exceptions to ISO 13485 that may apply to them, some of which are discussed in this article. As the standards evolve, it will become essential to evaluate the impact of the changes and whether this rule should be amended.
The new QMSR is structured according to the following numbering scheme:
Subpart A – General Provisions
820.1 Scope.
820.3 Definitions.
820.7 Incorporation by reference.
820.10 Requirements for a quality management system. (Note: This section links additional FDA requirements such as MDR, UDI, Corrections & Removals, and Tracking; applicability of Design and Development activities.)
Subpart B – Supplemental Provisions
820.35 Control of records. (Note: This section supplements record keeping activities, complaint/servicing records, UDI, and confidentiality.)
820.45 Device labeling and packaging controls.
What Is A Quality System?
In the comments the FDA received from the public, several are particularly insightful and warrant close attention. Notably, Comment 27 stands out for its foundational importance to the ethos of the QMSR, as it introduces the concept of “culture of quality,” which is paramount to compliance. Surprisingly, this comment was not positioned as the first, but it resonates with the core expectations of FDA:
…utilizing the definition in ISO 9000 for the term ‘‘top management’’ does not change that FDA expects medical device manufacturers, led by individuals with executive responsibilities, to embrace a culture of quality as a key component in ensuring the manufacture of safe and effective medical devices that otherwise comply with the FD&C Act.
A culture of quality meets regulatory requirements through a set of behaviors, attitudes, activities, and processes. Top management ensures that applicable regulatory requirements are met through the integration of QMS processes.
This comment provides an important moment to pause and consider FDA’s perspective on the importance of an organization’s quality culture. While adherence to processes and related activities is a cornerstone of compliance, FDA is acknowledging here that the creation of truly safe and effective products is inextricably linked to the demonstration of quality-centric ethos by top management. It is the embodiment of top management’s behaviors and attitudes, which filter throughout the fabric of the entire organization, that elevates quality’s importance to create truly safe and effective products.
Another pivotal statement connecting risk management and quality systems listed in Comment 19 states:
…Although the integration of risk management principles throughout ISO 13485 does not represent a shift in philosophy, the explicit integration of risk management throughout the clauses of ISO 13485 more explicitly establishes a requirement for risk management to occur throughout a QMS and should help industry develop more effective total product life-cycle risk management systems…
Before understanding this comment, the language and terminology must be explored.
In understanding the interplay between different definitions and terms, Comment 24 and the FDA’s response outline the integration of ISO 9000 terminology and definitions within the QMSR framework, specifically those delineated in Clause 3 as they are applied to ISO 13485:
“FDA considers the terms and definitions in ISO 9000 [Clause 3], as used in ISO 13485, to be incorporated by reference into the QMSR except for those terms and definitions FDA has determined are necessary to define in § 820.3 to satisfy requirements within the FD&C Act or its implementing regulations…This includes the corresponding notes for terms defined in ISO 9000, and as stated previously, FDA considers these notes as providing important context for understanding and implementing the standard rather than setting forth regulatory requirements…”
It follows that while the FDA has incorporated the terms and definitions from ISO 9000 (Clause 3) as utilized within ISO 13485 into the QMSR, it does not adopt them wholesale. The FDA reserves the right to define certain terms within 21 CFR Part 820.3 that are critical for complying with the mandates of the FD&C Act. ISO 13485 uses ISO 9000 as a normative reference and FDA’s definitions may vary from those in ISO 13485 and ISO 9000. ISO 13485 refers normatively to the terms and definitions in Clause 3 of ISO 9000, which the QMSR has adopted, and does not extend this reference to the entirety of the document; FDA considers the remainder of ISO 9000 to fall outside the scope of the QMSR. FDA considers the terms and definitions in ISO 9000 (Clause 3), as used in ISO 13485, to be incorporated by reference into the QMSR except for those terms and definitions FDA has determined are necessary to define in § 820.3 to satisfy requirements within the FD&C Act or its implementing regulations.
Moreover, in any instance where clauses of ISO 13485 conflict with those of the FD&C Act and/or its other implementing regulations, the latter will take precedence. A notable example is the definitions of “device” and “labeling,” in sections 201(h) and (m) of the FD&C Act, respectively, which supersede those in ISO 13485. Additionally, the FDA, in response to Comment 51, clarifies that the term “safety and performance” as mentioned in ISO 13485 is interpreted to align with “safety and effectiveness” in section 520(f) of the FD&C Act, ensuring a consistent approach.
ISO 13485:2016 clause 3.17 uses ISO 14971:2007 clause 2.16 as its definition of risk since the 2019 standard was not published in 2016. However, there are many definitions of risk in everyday life that may become confusing to a reader. For example, the Cambridge English Dictionary has over a dozen definitions of the term “risk,” and none of them reflect the definition used in the regulations, as risk is defined early in ISO 14971. This definition reads:
Risk: Combination of the probability of occurrence of harm and the severity of that harm
While it’s important to know that this definition of “risk” differs from the definition given in ISO 9000:2015, ISO 14971:2019 uses exactly the same definition as ISO 13485:2016 3.17. FDA confirms this understanding, and declines to create a new definition of risk, as shown in Comment 31:
“ISO 13485 Clause 0.2 states that ‘‘when the term ‘risk’ is used, the application of the term within the scope of this International Standard pertains to safety or performance requirements of the medical device or meeting applicable regulatory requirements. For these reasons, we do not believe that a definition for ‘‘risk’’ unique to the QMSR is necessary and are retaining the unmodified definition in ISO 13485.”
The Risk Spotlight
The Center for Devices and Radiological Health (CDRH) emphasizes risk and risk management in the newly published QMSR – much more than in the 1996 QSR. The focus represents an evolution from the original 1996 Quality System Regulation, although FDA maintains the emphasis on risk management concepts does not represent a significant departure from its foundational philosophy on risk. The FDA’s historical perspectives on risk management can be found in the preamble to 21 CFR 820:1996 Comment 83, a part of the document which is often overlooked. The construction of 21 CFR 820:1996 was based on ISO 9001:1994 and coincided with the first edition of ISO 13485, leading to the limited term “risk analysis” referring only to “Design Validation” – a reflection of the standards at that time. Of course, ISO 13485 progressed through its revisions to fully integrate risk management, while 21 CFR 820 was not revised. So, FDA had to rely on the preamble to convey its risk management intentions and await the future ISO 14971 being developed by ISO Technical Committee 210. ISO 13485 enriched the FDA and its investigators with a robust framework and regulatory language, particularly regarding risk related 483s. Fast-forward to today, and CDRH recognizes this shift to more explicit risk management with the implementation of QMSR. In response, the agency is training premarket and post-market personnel on both ISO 13485 and ISO 14971.
Expectations For Compliance With ISO 14971 With The Implementation Of QMSR
In comment 9 of QMSR, FDA explicitly states,
“…Aside from Clause 3 of ISO 9000, FDA does not, in this rulemaking, incorporate ISO 14971 or any other standards referenced by, or listed as a source in, ISO 13485, but acknowledges that these other standards may be helpful in understanding application of ISO 13485…”
While FDA felt it was legally important to add comment 9 to clarify the regulatory expectation for ISO 14971 compliance, it does clarify in comment 19 that:
“…Although the integration of risk management principles throughout ISO 13485 does not represent a shift in philosophy, the explicit integration of risk management throughout the clauses of ISO 13485 more explicitly establishes a requirement for risk management to occur throughout a QMS and should help industry develop more effective total product life-cycle risk management systems…”
While ISO 13485 does not expressly mandate ISO 14971 as a normative requirement and may not fully address risk as part of each and every clause, the FDA disagrees with the notion that ISO 13485 does not require a complete risk management system. ISO 13485 is intended to guide development of a QMS to meet regulatory requirements for medical devices, and it prioritizes that an effective quality system should systematically identify, analyze, evaluate, control, and monitor risk throughout the product life cycle to ensure that the devices are safe and effective.
What does this mean practically for industry? Compliance with every part of ISO 13485:2016 that explicitly discusses risk management is required and could be subject to regulatory inspection and consequences for noncompliance. The totality of the risk management system that wraps around and enhances all the risk-related clauses in ISO 13485 is highly suggested (by FDA) to be designed to comply with ISO 14971. If we think of this “suggestion” like we consider FDA “guidance” documents, we know it is a good idea to take this suggestion seriously.
Industry interpretation of ISO 13485 acknowledges the necessity of risk management and documentation within product realization (Clause 7.1). However, the standard also extends the requirement for risk consideration into other areas, such as:
- Quality management system (4.1.2), advocating a risk-based approach
- Human resources (6.2), stipulating risk-based personnel competency
- Purchasing (7.4), requiring controls that are proportionate to the risk of the device and also to the purchased product
- Monitoring and measurement (8.1), incorporating feedback into risk management
The language of the standard frequently invokes the concept of risk: “risk management” is used nine times in 13485; “risk-based approach” twice, and “risk” appears five additional times (outside the definitions).
The diagram below demystifies the product realization interactions between ISO 13485 and ISO 14971. We can even hazard to say that ISO 14971 can be the “sine qua non” of ISO 13485 and new QMSR compliance.
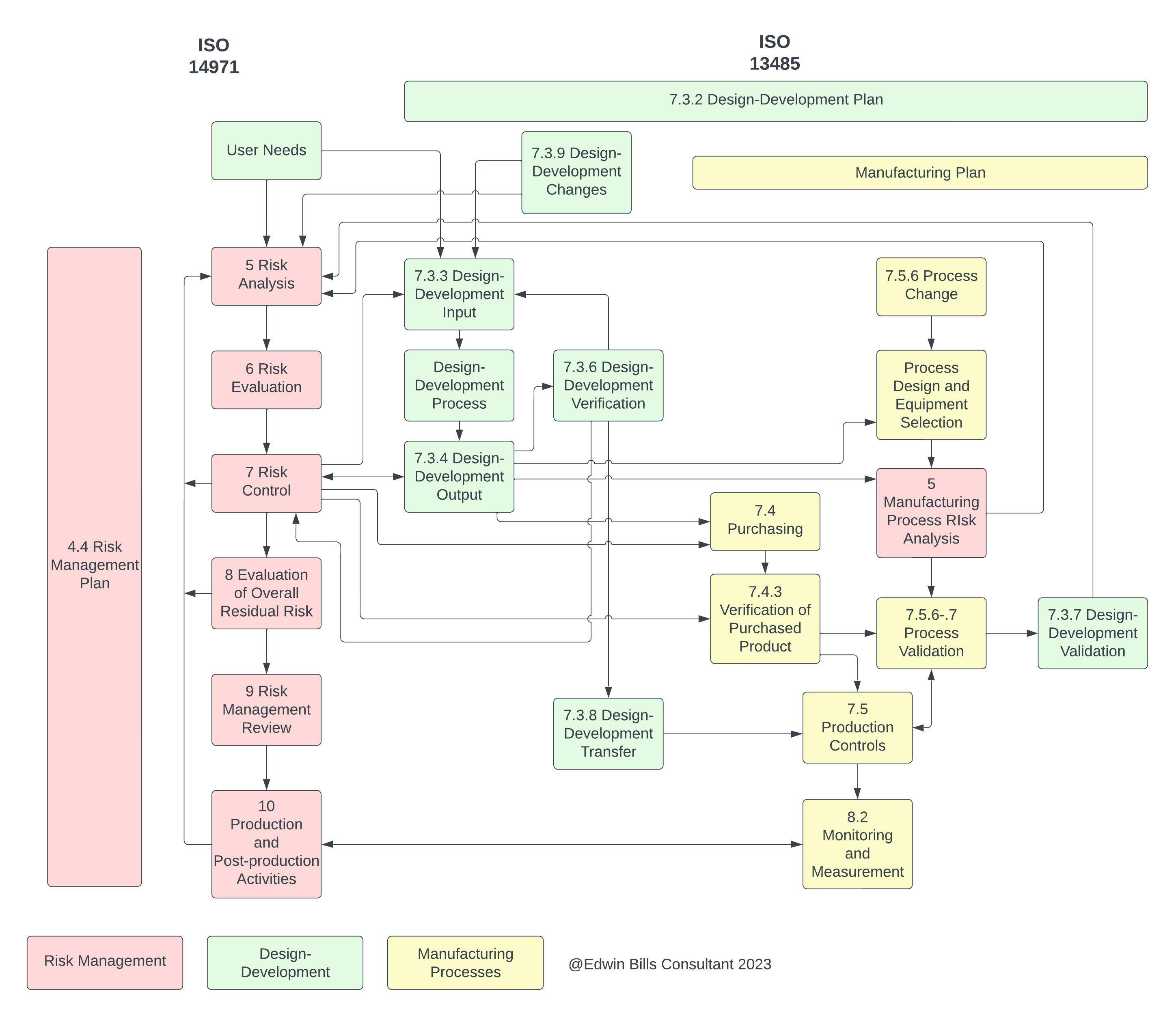
Click on image to enlarge.
A deep dive into product realization, specifically Design Development, Clause 7.3 of ISO 13485:2016 and its relationship to ISO 14971:2019, discusses what was erstwhile considered “design controls” by the FDA. The diagram above clarifies the interactions between various subclauses of 7.3 and ISO 14971:2019. Following from this, FDA further goes on to say in Comment 19:
“…In adopting ISO 13485, the QMSR incorporates risk management throughout its requirements and explicitly emphasizes risk management activities and risk-based decision making as important elements of an effective quality system …the ISO prioritizes that an effective quality system systematically identify, analyze, evaluate, control, and monitor risk throughout the product life cycle* to ensure that the devices they manufacture are safe and effective…”
*It is important to note that ISO 14971 provides a framework for managing risks throughout the entire product life cycle.
What To Do Next? P-D-C-A

Assuming that having an ISO 13485 certificate is equivalent to QMSR compliance is a dangerous thought for manufacturers and is not accepted by FDA. A thorough gap analysis should be done, especially with regard to interfaces and requirements of ISO 14971 with ISO 13485 as stated in this article.
One important note, and a common pitfall for medical device manufacturers, is this: completing an FMEA for the product is not enough to comply with the new QMSR (or with ISO 13485). FMEA, while a common and useful tool for later in the design process once design outputs are established, does not constitute the totality of holistic risk management. If you have a risk management file, and the only document analyzing risks is an FMEA, you have work to do. For more understanding of this concept, as well as decades of best practices all packed into one document, look at ISO/TR 24971:2020, Medical devices – Guidance on the application of ISO 14971.
A PDCA process as shown above can be followed with the following preliminary three steps for planning:
Plan, Do, Check, Act
- Read and understand the entire Federal Register article, including the comments and the regulation.
- Perform a gap analysis between your present quality system and the new QMSR.
- Plan: Develop a quality plan to address the gaps.
- Do: Execute the plan, including training the affected personnel.
- Check: Perform an internal audit to assure you have met the new requirements.
- Act: Implement the new quality system on Feb. 2, 2026.
Be careful! FDA has emphasized in their presentation at AAMI NeXus on Feb. 20, 2024, that the QMSR will not be effective until Feb. 2, 2026, and that they will be inspecting facilities to the present 1996 QMS until then. Any quality system changes to comply with the new QMSR should be carefully planned so as not to remove compliance with the old 21 CFR 820:1996 to avoid inspection issues.
Conclusion
The QMSR is intricately shaped by the principles of ISO 13485:2016. It retains the core of the old Part 820, selectively aligning with this globally recognized standard to ensure international consistency. Nonetheless, the FDA reserves the right to deviate from ISO standards when necessary to uphold the stringent requirements of the Federal Food, Drug, and Cosmetic Act or its implementing regulations.
Risk management is a key focus of the new regulation, with an emphasis on integrating risk management principles throughout the QMS. Despite strides toward global harmonization through initiatives like the Medical Device Single Audit Program (MDSAP), FDA maintains inspection authority and does not consider existing ISO 13485 certificates as proof of compliance with QMSR. Manufacturers need to fully understand the relationship between ISO 13485 and ISO 14971, with a focus on life cycle risk management starting from design to post-market to ensure full compliance with the new regulation that goes live in 2026. In alignment with this regulatory advancement, FDA is proactively training pre-market and post-market CDRH personnel on both ISO 13485 and ISO 14971. If you work in medical devices, this is your call to action: get trained on ISO 13485:2016 and ISO 14971:2019.
Recommended Further Readings
If you aren’t already familiar with the below ISO standards and other reading materials, we recommend reading them:
- The Intersection of ISO 13485 And ISO 14971 Under the Proposed FDA QMSR, by Edwin Bills and Christie Johnson, Med Device Online, May 24, 2023; accessed February 7, 2024; https://www.meddeviceonline.com/doc/the-intersection-of-iso-and-iso-under-the-proposed-fda-qmsr-0001
- ISO 13485:2016; https://www.iso.org/store.html
- A practical guide ISO 13485:2016, Medical devices, Advice from ISO/TC 210; https://www.iso.org/store.html (great resource on implementing 13485)
- ISO 9000:2015; Clause 3, Terms and Definitions (only) https://www.iso.org/store.html
- ISO 14971:2019; https://www.iso.org/store.html
- ISO TR 24971:2020; https://www.iso.org/store.html (must have)
- Federal Register/Vol. 89, No. 23/Friday, February 2, 2024, pp 7946-7525; Docket FDA-2021-N-0507-Final Rule
- 21 CFR 820 Amendment Final Rule, Presentation at AAMI NeXus 2024, February 20, 2024, Keisha Thomas, Kimberly Lewandowski-Walker, Washington, DC
- FDA Aligns U.S. Medical Device Quality System Regulation with International Standards, King and Spalding, Client Alert, February 8, 2024, https://www.kslaw.com/attachments/000/011/508/original/ca020824v7.pdf?1707499221 (another great resource)
 About The Authors:
About The Authors:
Edwin L. Bills, RAC, ASQ Fellow, CQE, CQA, CMQ/OE, has been a member of ISO TC 210 JWG1 for more than 20 years. This is the ISO group responsible for medical device risk management and the creation and maintenance of ISO 14971:2019, the risk management standard for medical devices, and ISO TR 24971:2020, the accompanying risk management guidance.
 Christie Johnson is a partner at Prodct LLC and director of quality at myBiometry. She is an advisor in product risk management and quality management systems to early-stage medtech startups and serves as a content expert to NIH's RADx initiative for the advancement of COVID-19 diagnostics. She was recently accepted into the ISO TC 210 committee for advancement of risk management standards for medical devices, ISO 14971:2019 and ISO/TR 24971:2020.
Christie Johnson is a partner at Prodct LLC and director of quality at myBiometry. She is an advisor in product risk management and quality management systems to early-stage medtech startups and serves as a content expert to NIH's RADx initiative for the advancement of COVID-19 diagnostics. She was recently accepted into the ISO TC 210 committee for advancement of risk management standards for medical devices, ISO 14971:2019 and ISO/TR 24971:2020.
 Jayet Moon earned a master’s degree in biomedical engineering from Drexel University in Philadelphia and is a Project Management Institute (PMI)-Certified Risk Management Professional (PMI-RMP). Jayet is also a Chartered Quality Professional in the UK (CQP-MCQI). He is also an Enterprise Risk Management Certified Professional (ERMCP) and a Risk Management Society (RIMS)-Certified Risk Management Professional (RIMS-CRMP). He is a Fellow of the International Institute of Risk & Safety Management. His new book, Foundations of Quality Risk Management, was recently released by ASQ Quality Press. He holds ASQ CQE, CQSP, and CQIA certifications.
Jayet Moon earned a master’s degree in biomedical engineering from Drexel University in Philadelphia and is a Project Management Institute (PMI)-Certified Risk Management Professional (PMI-RMP). Jayet is also a Chartered Quality Professional in the UK (CQP-MCQI). He is also an Enterprise Risk Management Certified Professional (ERMCP) and a Risk Management Society (RIMS)-Certified Risk Management Professional (RIMS-CRMP). He is a Fellow of the International Institute of Risk & Safety Management. His new book, Foundations of Quality Risk Management, was recently released by ASQ Quality Press. He holds ASQ CQE, CQSP, and CQIA certifications.